Title: An examination of some methods employed in determining the atomic weight of Cadmium
Author: John E. Bucher
Release Date: September 4, 2023 [eBook #71561]
Language: English
Credits: Richard Tonsing and the Online Distributed Proofreading Team at https://www.pgdp.net (This file was produced from images generously made available by The Internet Archive)
Transcriber’s Note:
New original cover art included with this eBook is granted to the public domain.
| I. | Introduction and Historical Statement | 1 | |
| II. | The Oxalate Method | 3 | |
| Preparation of Pure Cadmium | 3 | ||
| Preparation of Nitric Acid | 4 | ||
| Purification of Water | 4 | ||
| Purification of Oxalic Acid | 5 | ||
| Preparation of Cadmium Oxalate | 7 | ||
| Procedure | 8 | ||
| Results | 13 | ||
| III. | The Sulphide Method | 16 | |
| Preparation of Hydrogen Sulphide | 16 | ||
| Preparation of Nitrogen | 17 | ||
| Mode of Procedure | 18 | ||
| Results | 24 | ||
| Discussion of the Results | 24 | ||
| Discussion of the Method | 26 | ||
| IV. | The Chloride Method | 33 | |
| Preparation of Cadmium Chloride | 35 | ||
| The Filters | 48 | ||
| Analytical Process | 52 | ||
| Results | 57 | ||
| Discussion of the Results | 58 | ||
| V. | The Bromide Method | 69 | |
| Preparation of Cadmium Bromide and Hydrobromic Acid | 70 | ||
| Method of Analysis | 78 | ||
| Results | 80 | ||
| Discussion of the Results | 80 | ||
| VI. | Syntheses of Cadmium Sulphate | 82 | |
| Results | 90 | ||
| Discussion of the Results | 91 | ||
| VII. | The Oxide Method | 94 | |
| Results | 96 | ||
| Discussion of the Results | 97 | ||
| Determination of Error | 104 | ||
| Discussion of the Oxalate Method | 114 | ||
| VIII. | Other Methods | 119 | |
| IX. | Conclusion | 122 | |
The author wishes to acknowledge his indebtedness for advice and instruction to Professor Morse at whose suggestion and under whose guidance this work has been carried on. He also wishes to express his thanks for instruction to Professor Remsen in Chemistry, Professor Williams in Mineralogy, Dr. Ames in Physics and Mr. Hulburt in Mathematics.
The atomic weight of cadmium has been investigated by a number of chemists but the results obtained vary between wide limits. The work described in this paper was undertaken with the object of finding the cause of the discrepancy in some of the methods employed. A complete historical statement has been given by Morse and Jones, (Amer. Chem. Jour., 14. 261.) and it is only necessary, here, to give a summary for the purpose of reference:
| Ratio. | At. Wt. Cd. | ||
|---|---|---|---|
| 1818, | Stromeyer, | Cd : CdO | 111.483 |
| (Schweiggers Jour. 22, 336.) | |||
| 1857, | Von Hauer, | CdSO4 : CdS | 111.935 |
| 2 (Jour. f. Prakt. Chemie 72, 338.) | |||
| 1859, | Dumas, 1st series. | CdCl2 : Ag | 112.416 |
| 2d „ | CdCl2 : Ag | 112.007 | |
| (Ann. Chim. Phys. [3], 55, 158.) | |||
| 1860, | Lenssen, | CdC2O4 : CdO | 112.043 |
| (Jour. f. Prakt. Chem. 79, 281) | |||
| 1882, | Huntington and Cooke, | CdBr2 : AgBr | 112.239 |
| „ „ | CdBr2 : Ag | 112.245 | |
| (Proceedings Amer. Acad. 17, 28) | |||
| 1890, | Partridge, 1st Series | CdC2O4 : CdO | 111.816 |
| „ 2d „ | CdSO4 : CdS | 111.727 | |
| „ 3d „ | CdC2O4 : CdS | 111.616 | |
| (Amer. Jour. Sci.[3], 40, 377) | |||
| 1892, | Morse and Jones, 1st Method, | Cd : CdO | 112.0766 |
| 2d „ | CdC2O4 : CdO | 112.632 | |
| 1892, | Torimer and Smithy | CdO : Cd | 112.055 |
| (Zeit. f. Anorg. Chem. I. 364) | |||
In this summary as well as in the rest of this paper the following 3atomic weights are used:
| Oxygen | = | 16.00 |
| Sulphur | = | 32.059 |
| Carbon | = | 12.003 |
| Chlorine | = | 35.45 |
| Bromine | = | 79.95 |
| Silver | = | 107.93 |
“Cadmium met. puriss. galv. reduc”, obtained from Schuchardt, was used for preparing pure cadmium. It was heated to redness in a current of hydrogen which had been purified by washing with both acid and alkaline solutions of potassium permanganate. This treatment converted the metallic powder into a bar which could be distilled in a vacuum. The metal was then distilled 4nine times in the same manner that Morse and Burton, Amer. Chem. Jour. 12, 219, had distilled zinc. All distillations were made slowly except the last one, which was made quite rapidly.
Whenever pure nitric acid was required, it was purified by distilling against a platinum dish and collecting the distillate in a smaller one of the same metal. The nitric acid used was dilute and free from chlorine.
The water used in this work 5was purified by distilling twice from an alkaline solution of potassium permanganate, always rejecting the first part of the distillate. Whenever water was needed in the preparation of a pure compound e.g. cadmium oxalate, oxalic acid, cadmium nitrate, etc., it was subjected to the additional process of being distilled against a large platinum dish which was kept cool by placing ice inside it.
Commercial oxalic acid was heated with a fifteen percent solution of hydrochloric acid until 6all was dissolved. The solution was then warmed for twenty four hours. On cooling, crystals of oxalic acid separated out and these were washed with a little cold water to remove the mother liquor. They were then dissolved in hot ninety-five percent alcohol and allowed to crystallize slowly on cooling. The acid was next crystallized from ether in which it is only sparingly soluble. After this it was boiled with water until the odor of ethyl acetate had disappeared. Finally it was recrystallized three times from water and dried in the air at ordinary temperatures.
A weighed piece of cadmium was dissolved in nitric acid and the excess of acid evaporated off. The nitrite was then dissolved in a large quantity of water and an equivalent amount of oxalic acid in solution added. The oxalate separated in a few moments as a crystalline precipitate. It was collected on a porcelain filter and washed thoroughly to remove nitric acid and ammonium nitrate. A considerable amount of ammonium nitrate was formed during the solution of the cadmium in nitric acid. 8The oxalate was finally dried in an air-bath for fifty hours, at 150°C.
Enough cadmium oxalate for a determination was placed in a weighing tube which had been tared against a similar vessel and dried at 150°C. until the weight remained constant. It was then poured into a weighed porcelain crucible. The tube and its tare were now dried again at the same temperature to constant weight in order to avoid any error resulting from moisture being absorbed by the cadmium oxalate 9which adhered to the weighing-glass. The crucibles used in these determinations were arranged in the same manner as those employed by Morse and Jones in their work on this method. A small porcelain crucible on whose edge were placed three short platinum wires bent in the shape of the letter U, was placed in a larger porcelain crucible. The platinum wires prevented the lid from sticking to the crucible after heating and also allowed the products of decomposition to escape. The glaze was removed from the outside of the larger crucible with 10hydrofluoric acid to avoid sticking when heated to a high temperature. A second pair of crucibles arranged in the same manner was tared against the first one and in all cases treated like it. After the oxalate had been poured into the weighed crucible, it was decomposed by placing the crucible with its contents in a cylindrical asbestus covered air-bath, and slowly raising the temperature until the mass beams uniformly brown in color. In the last five determinations, the temperature was not allowed to exceed 300°C and after from forty to eighty hours 11the loss in weight was about ninety percent of the amount calculated for complete decomposition. In the first four the temperature was much higher and the time employed shorter. After the oxalate had been thus treated nitric acid was added and the contents of the crucible dissolved completely. The crucible was then transferred to a bath constructed by placing a larger porcelain crucible in a still larger one of iron and filling the intervening space with sand. It was slowly heated until the nitric acid had all evaporated and the dry nitrate began to give off red fumes. The crucibles 12were then removed to a similar bath containing iron filings instead of sand. This bath was heated by means of a single burner as long as red fumes were observed, and then for about five hours with a triple burner. Finally, the crucibles were transferred to a nickel crucible in the bottom of which a plate of unglazed porcelain was placed. The nickel crucible which had previously been set tightly into a hole cut in an asbestos board was then heated over the blast lamp for two hours. After this the porcelain crucible and contents were weighed and then reheated for half hour periods 13as before until three successive weighings remained constant. This usually required from three to four hours of blasting. In all determinations, the resulting product was tested for oxides of nitrogen with potassium iodide, starch and hydrochloric acid, but none was found. All weighings were reduced to the vacuum standard on the assumption of 8.4 for the Sp. Gr. of brass, 21. for platinum, 3.31 for the oxalate and 8.15 for cadmium oxide. The results are:
| Cadmium Oxalate. | Cadmium Oxide. | At. Wt. Cd. | |
|---|---|---|---|
| I | 1.97674 | 1.26414 | 111.73 |
| II | 1.94912 | 1.24682 | 111.82 |
| III | 1.96786 | 1.25886 | 111.77 |
| 14IV | 1.87099 | 1.19675 | 111.77 |
| V | 1.98941 | 1.27242 | 111.79 |
| VI | 1.37550 | .87994 | 111.85 |
| VII | 1.33313 | .85308 | 111.95 |
| VIII | 1.94450 | 1.24452 | 112.04 |
| IX | 2.01846 | 1.29210 | 112.09 |
A glance at these results shows that there is a variation of .36 of a unit and that the atomic weight in general increases with the number of determinations. In the first four determinations, there may have been loss of cadmium by reduction and subsequent volatilization, but in the later determinations this is not probable. It is believed that the greater part of the variation 15was due to imperfect dehydration of the oxalate. This and other sources of error in this method will be referred to later. The nickel crucible used gave a slight sublimate on heating, even after fifteen hours’ blasting. This condensed on the porcelain crucible as a brownish coating but, as both the crucible and its tare were blasted for the same length of time, it did not seem to change the difference of their weights. More than a dozen nickel crucibles were tried but none was found not to give a sublimate. The amount was so slight that no attempt was made to determine its nature.
This method is based on the conversion of cadmium oxalate into cadmium sulphide by heating in a current of hydrogen sulphide. The method has been used by Partridge. This result was 111.61 for the atomic weight of cadmium.
In the present work this gas was always prepared from potassium hydrosulphide which was made from barium sulphide (commercial). Barium sulphide was treated with dilute hydrochloric acid and the resulting hydrogen sulphide 17washed thoroughly with a solution of potassium hydrosulphide and then with pure water. It was then passed into a strong solution of potassium hydroxide until the later was saturated. When it was required, it was set free from this solution by adding dilute sulphuric acid and again washing the resulting gas with a strong solution of potassium hydrosulphide.
Whenever a current of nitrogen was required, it was prepared by passing air over a layer of hot copper gauze in a combustion 18tube. A short layer of copper oxide was first introduced, then the copper gauze and finally another layer of copper oxide. The air was dried with caustic potash before entering the tube and the nitrogen obtained was also passed through a long tube filled with lumps of this substance before being used.
A number of weighing tubes 140 millimetres long and 13 millimetres internal diameter were made especially for this work. They were always used in pairs, one being kept as a counterpoise. 19A porcelain boat of such dimensions as just to slide into the tube was placed in each one. For a determination, a tube and its boat were tared with another tube and boat, glass against glass and porcelain against porcelain until the difference in weight was less than two tenths of a milligramme. Both boats were heated in a current of hydrogen sulphide to incipient redness for about one hour. The current of hydrogen sulphide was then replaced by one of nitrogen, in which the boats were cooled, but while still warm they were transferred to their weighing tubes and allowed 20to cool in a dessiccator containing caustic potash and weighed. Before weighing, the stoppers of the weighing tubes were loosened for a moment in order to equalize the internal and external pressure. This treatment was usually repeated two or three times and the difference in weight remained perfectly constant. A portion of cadmium oxalate sufficient for a determination was placed in the weighed boat and dried at 150°C. The oxalate had been prepared exactly like that used in the oxalate method which has already been described. The gas pressure in the laboratory varied 21very much while this method was under investigation and great difficulty was experienced in maintaining a constant temperature although a thermoregulator was used. Sometimes a specimen of oxalate which was supposed to be dry would lose several tenths of a milligramme when the thermometer would only have gone up to 160°C or 165°C for an hour by accident. Under these conditions the drying was so uncertain that only four determinations were completed although many were started. The boat containing the oxalate which had been dried and weighed was 22placed on supports of unglazed porcelain in a combustion tube and a current of dry hydrogen sulphide passed over it. As soon as the air was expelled, the tube, which was in a combustion furnace, was slowly heated until all the oxalate seemed to be decomposed and then raised to dull redness. After this temperature had been maintained for about an hour, the sulphide was allowed to cool to a temperature of about 200°C. and the current of hydrogen sulphide replaced by dry nitrogen, using a three-way stopcock. When nearly cold the 23boat was slipped into its weighing-tube and weighed, the same precautions being used as when weighing the empty boat.
At this stage the sulphide was always from one to two milligrammes lighter than at the end of the determination. It was reheated for periods of one hour until the weight remained constant. This generally required from three to five hours. All weighings were reduced to the vacuum standard on the basis of 4.5 for the Sp. Gr. of cadmium sulphide, 3.31 for the Sp. Gr. of cadmium oxalate, 8.4 for the Sp. Gr. of brass weights and 21 for the Sp. Gr. of platinum weights.
24The results are as follows:
| Cd C2O4 | CdS | At. Wt. Cd. | |
|---|---|---|---|
| I. | 2.56319 | 1.84716 | 112.25 |
| II. | 2.18364 | 1.57341 | 112.17 |
| III. | 2.11643 | 1.52462 | 112.05 |
| IV. | 3.13105 | 2.25582 | 112.12 |
The first three determinations were made exactly as above described, the heating in hydrogen sulphide being done in a Bohemian glass combustion tube. The hydrogen sulphide was dried with calcium chloride.
The fourth determination was made under somewhat different conditions. The boat containing the weighed oxalate 25was placed in a combustion tube which passed through an asbestus covered air-bath. The air was displaced by a current of dry hydrogen sulphide and the bath slowly heated. When the temperature had risen to 210°C. it was maintained there for three hours, and then raised to 250°C. for three hours. The sulphide then weighed 2.27 grammes, being 14 milligrammes heavier than when the determination was finished. It was replaced in the tube and reheated in a current of hydrogen sulphide at a temperature of 300°C. for four hours. It was then transferred to a porcelain tube and heated to 26redness for one hour. It then weighed 2.25437 grammes, being 1.45 milligrammes lighter than at the end of the determination. The weight did not become constant until it had been heated six hours more to redness in a current of hydrogen sulphide. When this oxalate was slowly heated in H2S, a small amount of oxalic acid sublimed to the colder part of the tube, but, in the other cases where the heating was more rapid, only carbon monoxide, carbon dioxide, and water were observed.
When hydrogen sulphide is passed 27through a red-hot tube, sulphur is deposited on the colder parts because at this temperature hydrogen sulphide dissociates and the elements do not recombine on cooling. In this work, a faint sublimate was noticed before coming to the zone of sulphur deposit. On exposure to air, it deliquesced in a few minutes forming small yellow drops which had a saline taste, and gave tests for potassium and sulphur. The sublimate had a yellow color and was evidently formed by the action of sulphur on glass. It seemed to do no harm, but in the fourth determination an effort was made to avoid 28it by using a porcelain tube instead of a glass combustion tube for heating to redness in a current of hydrogen sulphide.
The fact that sulphide of cadmium was always too light after the first hour’s heating in hydrogen sulphide proves that it must have contained some oxide of cadmium even after this heating. Oxide of cadmium is readily absorbed by the glaze on porcelain, and some error must have been introduced in this way because it would not be converted into sulphide after forming a silicate.
The effect of this would be to give a low result for the atomic weight of cadmium. To get some 29idea of the magnitude of this error, the sulphide was poured out of the boats used in the first and second determinations. They were then warmed with nitric acid for a few moments, washed in water, and heated over the blast lamp for a few minutes. The boats used as tares were treated in exactly the same manner. On weighing, the boats in which the oxalate in determinations I and II had been decomposed, were found to be 1.12 milligrammes and .82 milligrammes heavier respectively than at the beginning of the determinations. This would only introduce an error of .03 of a unit in the 30atomic weight on account of the small difference in weight between these amounts of oxide and equivalent amounts of sulphide. The boats were warmed, as above mentioned, with nitric acid to remove any adhering sulphide. This might have decomposed some cadmium silicate at the same time, and the error due to cadmium oxide thus be found smaller than it really is.
The following experiment was made in the hope of avoiding the formation of cadmium silicate. The glaze was removed from the inside of a porcelain boat by hydrofluoric acid followed by a thorough scouring with sand and 31water. The boat was then heated in the flame of a blast lamp for several minutes, tared against another boat which was not treated with hydrofluoric acid. Both were heated to redness in a current of hydrogen sulphide for an hour, cooled, weighed, and then heated in hydrogen sulphide for another hour, and weighed again. The boat gained 1.7 milligrammes during this second heating, showing that a boat whose glaze has been removed by hydrofluoric acid could not be used in this method. Throughout this work, great care was taken to exclude 32the oxygen of the air from the cadmium sulphide, while hot. The current of hydrogen sulphide in which the cadmium sulphide is heated must not be too slow, otherwise the sulphur in the dissociated gas will diffuse to the colder parts of the tube and condense, the residual gas becoming very rich in hydrogen. The hydrogen will then reduce some of the sulphide to metal, causing loss by volatilization. One determination was lost in this way, over two milligrammes of the sulphide being sublimed out, and it could easily be detected on the side of 33the tube. It is believed that the cause of the variations in the four determinations made by this method, is due to imperfect dehydration of the oxalate. It did not seem advisable to continue this part of the work any farther; therefore the chloride method was taken up.
Huntington had determined the ratios of CdBr2 to AgBr and also CdBr2 to Ag very carefully, obtaining the result 112.24 for the atomic weight of cadmium. Morse and Jones had obtained 112.07 for this 34constant by the oxide method. The object of the work about to be described was to find the cause of this discrepancy if possible. It was thought advisable however to make some determinations of the ratio of CdCl2 to AgCl before beginning the bromide method.
Dumas, in 1859, used cadmium chloride to determine the atomic weight of the metal. He did not establish its ratio to silver chloride but to silver by titration. He prepared cadmium chloride by dissolving the metal in hydrochloric acid and melting the resulting product in a platinum capsule 35for five or six hours. He made two series of three determinations. The chloride used in the first series was yellow in places and not completely soluble. The result was 112.476. The second series was made with chloride which was perfectly white and soluble and gave 112.007 for the atomic weight of cadmium. It is evidently more reliable than the first series and Dumas himself concluded that the atomic weight is very near 112.01.
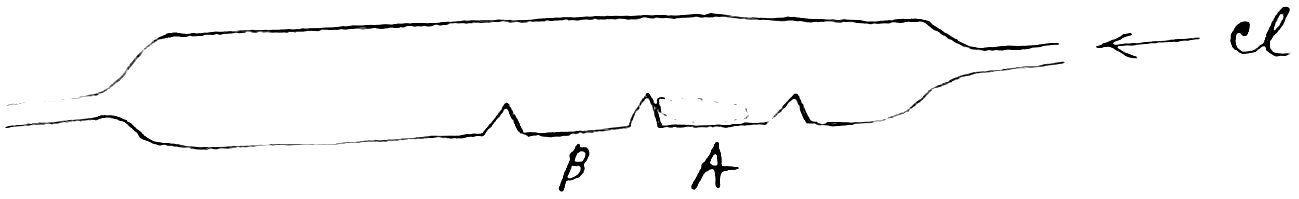
Four different specimens of cadmium chloride were used in this work and from these 36specimens portions were taken for analysis. These portions were treated differently in different analyses, therefore it will be necessary to give a brief descriptions of them and mention the number of the determinations, in which each one was used. Chloride of cadmium was prepared in the following manner. A solution of pure hydrochloric acid was prepared by passing a current of hydrochloric acid gas into pure water which was contained in a porcelain crucible until no more was absolved. The water used had been purified by distilling against a platinum dish and the 37hydrochloric acid gas was obtained by heating ordinary concentrated chemically pure hydrochloric acid in a distilling bulb whose neck had been closed by fusion in order to avoid the use of a cork or rubber stopper. Hydrochloric acid thus prepared will leave no residue on evaporation when air is excluded (Stas, Aronstein’s German translation, p. 111). A piece of platinum foil freed from iron by heating in the vapors of ammonium chloride as recommended by Stas (Aronstein’s translation, p. 112) was introduced and a piece of cadmium laid on it. Solution begins at once, the hydrogen being liberated on 38the platinum foil. During the later part of the process, heat was applied. After all of the metal had dissolved, the solution was evaporated, the platinum foil having previously been removed. The crystals of cadmium chloride which separated were not dried but allowed to remain slightly moist with hydrochloric acid. If no platinum foil is used, the solution of the pure metal becomes exceedingly difficult, unless a very large excess of acid is used. No objection can be raised to the use of platinum foil for in making fifty grammes of cadmium chloride it cost less than a tenth 39of a milligramme and even this could probably have been avoided by using a somewhat larger amount of hydrochloric acid. The foil was always kept submerged in the acid liquid. The moist crystals of cadmium chloride were transferred to a combustion tube passing through an asbestus covered air-bath, and dried in a current of hydrochloric acid gas for several hours at 300°C. The hydrochloric acid gas had been passed through a long calcium chloride tube to dry it, although calcium chloride probably does not do this very thoroughly. The hydrochloric acid gas was then replaced by 40a current of nitrogen prepared as has already been described under the sulphide method. After the current of nitrogen had been passing for about half an hour, the tube was allowed to cool, and the chloride transferred to another combustion tube, one end of which had been sealed in the flame of a blast lamp. The other end was drawn out and attached to a Sprengel mercury pump. After exhausting, the chloride was sublimed in the vacuum. This takes place at a moderate temperature and the sublimate has a beautiful crystalline structure and is perfectly white. 41The crystalline mass exposes so much surface that water is taken up very rapidly when exposed to the air. This action is so rapid that the crystals cannot be transferred to a weighing-glass without introducing an appreciable error. The whole sample was accordingly transferred to a stoppered glass bottle which was kept under a bell jar with sticks of caustic potash. Three samples were prepared in this manner, the first being used in determination one, the second in determinations two to seven inclusive, and the third in determinations eight to nineteen inclusive. 42The samples used in determinations twenty and twenty-one were prepared in the following manner: About three grammes of cadmium were placed in a combustion tube in which three bridges (as in the distillation of pure cadmium) had been made. A section may be represented thus
The metal was placed in cavity A and a stream of chlorine passed through the tube. The chlorine was prepared from potassium bichromate and hydrochloric acid and dried 43by passing it through a long tube containing calcium chloride. When the air had been displaced, the cadmium was heated. It fused and began to burn to the chloride which partly flowed over the bridge into cavity B and partly distilled over into this cavity. When the reaction had ended, the current of chlorine was replaced by one of dry nitrogen, and the tube was allowed to cool and the chloride taken for analysis XX. The specimen used in analysis twenty-one was prepared in exactly the same way, only the chlorine used was obtained from manganese dioxide, sodium chloride 44and sulphuric acid, and was dried with phosphorous pentoxide instead of calcium chloride.
The special treatment of the portions taken for analysis was as follows: Those taken for determinations I, II and from XI to XIX inclusive were placed in a platinum boat and put into combustion tube. A current of hydrochloric acid gas obtained by heating the aqueous acid was passed through the tube. The gas had been dried by calcium chloride. When the air was displaced, the chloride was heated somewhat higher than its fusing-point 45i.e. to incipient redness, and maintained there for a length of time varying from a few minutes to more than an hour. The hydrochloric acid was then displaced by a current of nitrogen, and the chloride allowed to cool. The boat with the chloride, while still slightly warm, was placed in a weighing-tube, cooled in a dedicator containing caustic potash and weighed. The chloride thus prepared is transparent and presents only a small surface to the air. It takes water up so slowly that no error is introduced from this source. This was tested in one case by 46allowing a boat containing some chloride thus prepared to stand in the air for a certain length of time and noting the increase in weight. It was quite slow. In several cases specimens of chloride were tested for hydrochloric acid using tropaeolin as an indicator. It was always found neutral. The portions used for determinations III and VI to X inclusive were prepared in exactly the same manner as the preceding ones except that the hydrochloric acid gas in which they were fused was not dried but used just as it came from 47the aqueous acid. In some cases the platinum boat in which the chloride was fused was weighed before and after the fusion. The weight remained unchanged.
For determinations IV and V, about six grammes of cadmium chloride were placed in a platinum boat, and more than two-thirds of it distilled out in a current of hydrochloric acid gas which had not been dried. Part of the distillate was collected after cooling in nitrogen and used in determination IV while the residue remaining in the boat was used for determination V. 48The method of preparing the chloride used in determinations XX and XXI has already been described.
Thinking that a Gooch crucible with a platinum sponge on the bottom in place of asbestus would be desirable for this work one was accordingly made and answered the purpose very satisfactorily. All determinations were made by using such filters. C. E. Munroe (Chem. News, Vol 58, p. 101) has described the preparation of these filters. 49A platinum Gooch crucible was placed on a filter paper and some ammonium platonic chloride which had been thoroughly washed introduced by suspending it in alcohol and then pouring this into it. The precipitate settles to the bottom forming a uniform layer and the alcohol drains, off through the filter paper. The crucible was then dried slowly in an air-bath. After this it was transferred to a porcelain crucible and slowly heated until decomposition was complete. In this manner a layer of platinum felt is obtained which acts as a very efficient 50filter. Another layer of double chloride was then decomposed as before so that if there were any imperfections in the first layer they would be covered by the second layer. The surface was smoothed down by means of a glass rod. To prepare a good filter the drying and subsequent heating should be very slow. The heating must not be at too high a temperature, otherwise the felt becomes very compact and is useless for filtering purposes. Pressure produces the same effect. The filters were always treated with strong nitric acid, washed and 51reheated before being used, but in no case was chlorine detected in the nitric acid after the washing, nor any loss in weight of the crucible. An objection to the use of these crucibles for the purpose named was found in the course of this work, but it will be discussed later. The crucibles were always set in a large weighing-glass, and another weighing-glass containing an equal amount of platinum foil used as a tare, in weighing. This precaution was perhaps unnecessary, but at least it did no harm.
The weighed cadmium chloride was dissolved by placing the boat containing it in an Erlenmeyer flask containing water. The boat was then washed, dried and replaced in its weighing-tube. On weighing again, the loss in weight is equal to the weight of cadmium chloride taken. All samples gave a perfectly clear solution except those used for determinations XX and XXI. A drop of nitric acid (1:3) was added to each solution except in determination XIV where the cubic centimetres 53were added, and in XVI where ten cubic centimeters were added. A solution of silver nitrate was then added to precipitate the chlorine. This as well as the subsequent washing was done in a dark-room illuminated by a single gas light whose rays had to pass through a strong solution of neutral potassium chromate. The precipitate was contracted by warming on the water-bath. It was then collected in the prepared Gooch crucibles and washed. Before filtering, the flask containing the precipitate and mother-liquor was 54allowed to cool. Silver chloride is soluble in water to a considerable extent but is reprecipitated by adding an excess of either silver nitrate or hydrochloric acid. Stas (Ann. de Chem. et Phys. [4], 25, 22; [5], 3, 145; [5], 3, 289.) investigated this very thoroughly. Cooke also did some work on it and used a dilute solution of silver nitrate to wash the chloride thus preventing solution (Proc. Amer. Acad. 17, 7.). In the above work, therefore, a solution containing 0.10 grammes of silver nitrate per liter was first used, followed by one only one-tenth as strong, and finally pure water was used. 55Only two or three washings could be made with water as the chloride went into solution after this owing to the removal of the silver nitrate. The last silver nitrate solution used is so weak that any error introduced by not washing it out completely is insignificant. After washing, the silver chloride was dried at temperatures varying from 150°C. to 300°C. to constant weight. A glass air-bath was used in order to prevent products from the burning gas from coming in contact with the chloride. It was then weighed. 56The quantity of silver nitrate used in the determinations was varied very much. The excess over what was required to precipitate the chloride is given in the table of results in those cases in which it is known. The quantity of water used in each determination is also given where it is known. It is given in the number of cubic centimetres used per gramme of cadmium chloride and does not include wash water. All weighings are reduced to the vacuum standard on the basis that Sp. Grs. of CdCl2 = 3.94 and AgCl = 5.5. The results are:
| 57 | ||||||
| No. | CdCl2 | AgCl | H2O per Grm. | Excess AgNO3 | At. Wt. | Melted in |
|---|---|---|---|---|---|---|
| I | 3.09183 | 4.83856 | 112.339 | Dry HCl | ||
| II | 2.26100 | 3.53854 | 112.329 | „ „ | ||
| III | 1.35729 | 2.12431 | 112.320 | Moist HCl | ||
| IV | 2.05582 | 3.21727 | 112.339 | „ „ | ||
| V | 1.89774 | 2.97041 | 112.306 | „ „ | ||
| VI | 3.50367 | 5.48473 | 8.90 | 112.283 | „ „ | |
| VII | 2.70292 | 4.23087 | 200 | 1.79 | 112.301 | „ „ |
| VIII | 4.24276 | 6.6398 | 300 | 8.10 | 112.387 | „ „ |
| IX | 3.40200 | 5.32314 | 300 | 18.95 | 112.368 | „ „ |
| X | 4.60659 | 7.20386 | 300 | 25.62 | 112.472 | „ „ |
| XI | 2.40832 | 112.434 | Dry HCl | |||
| XII | 2.19114 | 3.42724 | 112.433 | „ „ | ||
| XIII | 2.84628 | 4.45477 | 300 | 4.45 + 3cc. HNO3 | 112.319 | „ „ |
| XIV | 2.56748 | 4.01651 | 300 | .10 | 112.399 | „ „ |
| XV | 2.31003 | 3.61370 | 300 | .10 + 10cc. HNO3 | 112.406 | „ „ |
| XVI | 1.25008 | 1.95652 | 300 | 4.66 | 112.319 | „ „ |
| XVII | 1.96015 | 3.06541 | 300 | 3.22 | 112.466 | „ „ |
| XVIII | 2.29787 | 3.59391 | 300 | 4.27 | 112.448 | „ „ |
| 58XIX | 1.94227 | 3.03811 | 300 | 3.61 | 112.423 | Dry HCl |
| XX | 1.10976 | 1.73547 | 112.471 | „ „ | ||
| XXI | 1.63080 | 2.55016 | 112.476 | „ „ | ||
| Average | 112.383 | |||||
In the first five determinations, the analytical operations were conducted as nearly as possible alike, but the preparation of the portions of cadmium chloride taken for analysis was varied very much as will be seen by referring back to this part of this paper. The results do not vary more than ±0.015 from their average. This is very 59strong evidence of the purity of the chloride used for, if it contained any impurity, we should have expected to vary the amount in the different portions. After this, attention was paid especially to the analytical process, for it was thought that there probably was some serious error in the method, the result being higher than any that had previously been obtained, if we exclude Dumas’ first series which he himself did not accept. The conditions were varied in many ways to see how much the result could be influenced, but under no conditions were 60results as low as Huntington’s average (112.24) obtained. A number of errors were found in the method during the work, but they seem to neutralize each other to a great extent. The more important ones will now be given. Nearly every filtrate including the corresponding wash water was examined for chlorine after the silver and cadmium had been precipitated by hydrogen sulphide. The excess of hydrogen sulphide was expelled by boiling, after the addition of some nitric acid. In two cases an inverted condenser was used. On adding silver 61nitrate a precipitate was always obtained showing the presence of chlorine. Care was always taken to filter off sulphur formed by the oxidation of hydrogen sulphide, before adding the silver nitrate. The precipitate was never very heavy, and was not estimated quantitatively. It is evident that cadmium nitrate exerts a solvent action on silver chloride. In some cases a very large excess of silver nitrate was added but it did not change the results markedly. Silver nitrate itself dissolved silver chloride to some extent. The increase in insolubility, if any, on adding 62an excess of silver nitrate is probably counterbalanced by the increased error due to occlusion of nitrates in the silver chloride. Stas (Aronstein’s Trans. p. 156) says it is impossible to contract silver chloride or bromide in a solution containing salts without there being occlusion and that the precipitate can only be freed from them by dividing up the contracted mass by shaking with pure water. This was not done here owing to the solubility of silver chloride in pure water, and the complications introduced in the analytical part. The occlusion 64of nitrates by the silver chloride would lower the atomic weight found. The silver chloride obtained always darkened on heating and contained cadmium, as was shown in the following manner: The lump of silver chloride was attached to the negative pole of a cell and electrolyzed in a bath containing dilute sulphuric acid. The resulting metal was then dissolved in nitric acid and the silver precipitated by adding hydrochloric acid. The filtrate was evaporated to expel the nitric acid and the residue taken up with water 65and tested for cadmium with hydrogen sulphide. An appreciable quantity was always found. This method of examination does not show the occluded silver nitrate. Another error which tends to lower the atomic weight found is due to the platinum crucibles used for filtering. If a silver nitrate solution is filtered through such a crucible there will be an increase in weight due to silver being deposited. This takes place in acidified solutions as well as in neutral ones. Washing with ammonia does not remove the deposit, but 66strong nitric acid does, the washings giving a test for silver. Whether the depositing of silver is due to the action of spongy platinum in contact with the compact metal of the crucible or to some impurity in the platinum sponge was not determined, but the former seems by far the most probable. The increase in weight during the time required for filtering a determination must have been quite small however. The samples of cadmium chloride employed for determinations XX and XXI were prepared by burning 67cadmium in a current of chlorine. The glass tube used was attached somewhat and the solution of the chloride was very slightly turbid in each case. The turbidity was so slight however, that no very serious error could have resulted from it, particularly as it was probably partly counterbalanced by the formation of some potassium chloride. For more accurate work, it should have been made and redistilled in a porcelain tube. These two samples were tested for free chlorine with potassium iodide and starch paste, but none was found. Some 68of the specimens of chloride prepared by fusion in a current of hydrochloric acid were found to be neutral, using tropaeolin as an indicator.
As nearly as can be judged, the above errors would probably counterbalance each other to a great extent, and thus give a fairly close approximation to the atomic weight of cadmium when the average of all the determinations is taken. The value 112.383 thus obtained can only be regarded as tentative.
Huntington (Proc. Amer. Acad. 11.28) working under the direction of J. P. Cooke, determined the ratio of cadmium bromide to silver bromide and using the total quantities for the calculation the result for the atomic weight of cadmium is 112.239. He also determined the ratio of cadmium bromide to silver, obtaining 112.245 for the atomic weight of cadmium.
The work which will now be described was carried out very much like the work described under the chloride method. The ratio of cadmium bromide to silver 70bromide was investigated.
A large quantity of hydrobromic acid was prepared according to the method described by Dr. Edward R. Squibb (Trans. of Med. Soc. of the State of N. Y.). One part of water was added to seven parts of strong sulphuric acid (Sp. Gr. = 1.83) and the mixture cooled. Then six parts of potassium bromide were dissolved in six parts of hot water and the diluted sulphuric acid added to this hot solution. It was set aside until 71cold in order to allow the sulphate of potassium to crystallize out. The crystals were drained on a filter-plate and quickly washed with two parts of water. The mother-liquor and washing were then distilled until no more acid was obtained on further heating. The acid thus obtained was distilled three times from potassium bromide, twice from cadmium bromide formed by adding a piece of pure cadmium to it, and twice without the addition of anything. It was tested and found to be free from sulphuric acid. Cadmium bromide was prepared 72from it, in exactly the same way that the cadmium chloride used in the chloride method was prepared from pure metal and hydrochloric acid. While the crystalline mass of cadmium bromide was still moist, it was transferred to a combination tube and dried at 300°C for several hours in a current of nitrogen. It was then sublimed in a vacuum as the chloride had been. This specimen served for the first three determinations. About nine grammes of it was placed in a platinum boat in a combustion tube, and part of it distilled 73in a current of nitrogen. The distillate, a portion of which had been tested with tropaeolin and found neutral, was used for determination I. The residue in the boat was used for determination II. Another portion of the main sample was resublimed in a vacuum and used in determination no. III. Cadmium bromide is not hygroscopic or at least only slightly, therefore the sublimed cadmium bromide can be transferred to a weighing-glass without taking up water. This cannot be done in the case of the chloride. 74It is probable that the hydrobromic acid as above prepared was perfectly free from hydrochloric acid. Chlorine in cadmium bromide would cause the atomic weight to be found lower than it really is. It was thought desirable, however, to prepare an acid which would certainly be free from chlorine. The method described by Stas (Aronstein’s German translation, p. 154.) was employed with the additional precaution that the above purified acid was used to start with and all reagents employed had been especially prepared so as to be free from chlorine. Pure silver was prepared 75according to Stas’ description (see Aronstein’s translation, page 34, also page 104) by the action of ammonium sulphite on an ammoniacal solution of silver nitrate and copper sulphate. The silver was dissolved in nitric acid free from chlorine, and then slowly added to a dilute solution of the above-described hydrobromic acid, and the precipitated silver bromide thoroughly washed. It was then digested for a long while in a strong solution of potassium bromide, first in the cold, then by heating. The potassium bromide had been made thus: 76Twice recrystallized potassium hydrogen tartrate was heated in a platinum dish in a muffle furnace until it was converted into carbonate, and the excess of carbon burned off. It was then dissolved in water, filtered and neutralized with some of the hydrobromic acid already described. The carbonate had been tested for both sulphuric acid and chlorine with negative results. After the silver bromide had been digested with the potassium bromide, it was washed very thoroughly, suspended in water, and a current of hydrogen sulphide passed into it. This converts it into sulphide 77hydrobromic acid being liberated. The acid was drained off on a porcelain plate, and then distilled a number of times. It was finally tested and found to be perfectly free from sulphates and also did not contain free bromine. Having started with an acid which was probably pure and subjected it to these operations with reagents free from chlorine, there can be no doubt as to the purity of the resulting acid. The hydrogen sulphide used was prepared from potassium hydrosulphide as in the sulphide method, and washed first with a solution of the hydrosulphide, then very thoroughly 78with pure water. From the hydrobromic acid obtained, a specimen of cadmium bromide was prepared as before and sublimed twice in a vacuum. This specimen was used for determinations IV and V.
The first three determinations were made exactly like those in the chloride method. The last two were also made in the same manner, only the washing of the precipitate was varied. After the silver bromide had been contracted by warming on a water-bath 79it was washed by decantation and then agitated violently with cold water to remove occluded nitrates, but it was then so finely divided that it could not be filtered. The artifice used by Stas to contract it a second time was to pass a current of steam into the milky liquid. This was tried here, but for some reason or other did not work very well, and considerable difficulty was had in filtering it. The results of the five determinations are tabulated below. All weighings are reduced to the vacuum standard on the basis of Sp. Gr. of CdBr2 = 4.6 and Sp. Gr. AgBr = 6.62.
| 80 | ||||||
| No. | CdBr2 | AgBr | H2O | Ex. AgNO3 | At. Wt. | Remarks |
|---|---|---|---|---|---|---|
| I | 4.39941 | 6.07204 | 112.43 | Distillate } | ||
| II | 3.18030 | 4.38831 | 112.42 | Residue } | ||
| III | 3.60336 | 4.97150 | 112.45 | Resublimed. | ||
| IV | 4.04240 | 5.58062 | 112.29 | |||
| V | 3.60505 | 4.97519 | 112.38 | |||
| Average | 112.394 | |||||
The first three specimens were prepared under widely different conditions yet the results agree quite closely. The last two were prepared from the repurified hydrobromic acid. If chlorine had been removed during the second purification we should expect a higher result 81but the results are lower. There seems to be hardly any doubt that this is due to analytical errors rather than a change in the composition of the bromide. Whether this be true or not, the five determinations all fall within the limits obtained by the chloride method and confirms it as fully as can be expected.
The errors of the method are the same as those of the bromide method, only they are probably less in most cases. One filtrate was examined for bromium, but none was found showing the method to be more perfect in this respect.
It was next thought of examining the method based on the conversion of cadmium sulphate into cadmium sulphide, which has been used by von Hauer whose result is 111.94 for the atomic weight of cadmium, and more recently by Partridge who obtained a much lower result, namely 111.73. They dried cadmium sulphate in porcelain boats, and then reduced it to sulphide by heating in a current of hydrogen sulphide. The reduction begins in the cold and is probably complete or at least nearly complete before the temperature is sufficiently high 83for cadmium sulphate to decompose into cadmium oxide, for the sulphate is very stable with respect to heat. This being the case, probably no error results from the formation of a silicate of cadmium in this method. The main difficulty in this method would be to prove that the cadmium sulphate used is free from water. Neither von Hauer nor Partridge has done this because drying a substance to a constant weight is not sufficient evidence of its anhydrous character, especially if the drying is done at a constant temperature. This has been shown very clearly in the case 84of copper sulphate, by Richards (Proc. Amer. Acad. Sci. 26. 263.)
It was therefore decided to attempt the synthesis of cadmium sulphate, hoping to be able to fix a minimum value for the atomic weight of cadmium.
A piece of hard glass tube was closed at one end by fusion and the other end drawn out into a small tube which was then bent twice at right angles. The large part was cut off near the beginning of the smaller tube, and the edges rounded by fusion. It was filled with dilute sulfuric acid and heated for some time to remove soluble 85matter from the glass. After removing this acid, a weighed piece of cadmium was introduced and an excess of dilute sulphuric acid (1: 3) added. The tube contained a small piece of platinum to aid the solution of the cadmium. During the process of solution, the two parts of the glass tube were held together by a rubber band, and the outlet of the smaller tube dipped under pure water contained in a small tube closed at one end.
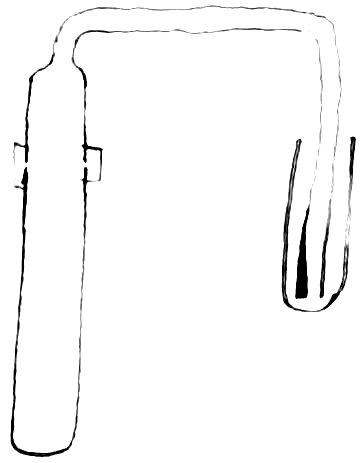
Fig. 2.
A section of the arrangement is shown in figure 2. Solution was aided by the application of heat. 86These precautions in dissolving the metal were taken to prevent loss by spraying. After the metal had been dissolved, the solution and the water through which the hydrogen had escaped were transferred to a porcelain crucible. An equal amount of sulphuric acid was then added to the tare and both were heated until fumes of sulphuric acid ceased to come off. The crucible containing the dry sulphate was next placed an a porcelain plate in a nickel crucible set in a hole in an asbestos board. This was placed over the flame of a Bunsen burner, so that the bottom of the nickel crucible was 87barely at a red heat. The temperature on the inside of this bath was considerably lower. After the weight had become nearly constant, the sulphate was tested for sulphuric acid by means of standard alkali using tropaeolin as an indicator. It was found acid, but so slightly that no attempt was made to estimate it. Result = 112.35 is preliminary.
Another synthesis was made as follows: A platinum crucible, lid and perforated cone were place in a large weighing-glass and tared with a similar weighing-glass containing a platinum crucible, platinum foil being added until the weights were 88equal. After these had been accurately weighed, a weighed piece of cadmium was added to the one containing the cone. The cone was inverted over the piece of metal on the bottom of the platinum crucible. A considerable excess of dilute (1: 3) sulphuric acid was then added, the lid whose edge was bent down placed on the crucible, and the weighing-glass stoppered loosely. This was placed in an air-bath, and gently warmed during the later part of the process of solution. There is no difficulty in getting complete solution if a sufficient excess of acid 89is used. A vertical section of the crucible and weighing-glass is shown in figure 3.
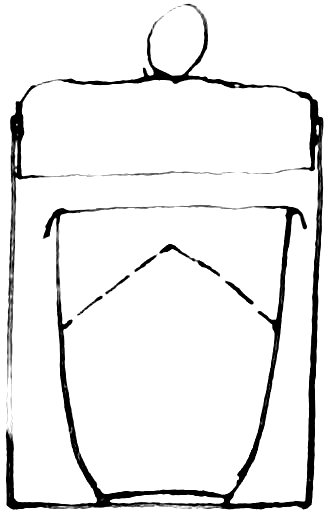
3
This arrangement avoids loss from spraying, and the necessity of transferring the solution from a tube to a crucible as in the first experiment.
An equal quantity of sulphuric acid was added to the crucible used as a tare and evaporated. After the metal had been dissolved, the platinum cone was lifted to one side and the excess of acid evaporated off. It was then heated in a glass air-bath for a long time at a temperature 90which was probably about 400°C. After the weight had become constant, the amount of free sulphuric acid was estimated by titration with a standard alkali using tropaeolin as an indicator. 1.25 milligrammes were found and this weight was subtracted from that found at the balance. Weighing were reduced to the vacuum standard, assuming the Sp. Grs. of cadmium and anhydrous cadmium sulphate[1] to be 8.54 and 3.0 respectively. The results were as follows:
1. Could not find any record of its Sp. Gr., 3.0 is assumed.
| Cd | CdSO4 | At. Wt. | |
|---|---|---|---|
| I. | (112.35 can only be regarded as a preliminary experiment) | ||
| II. | 1.15781 | 2.14776 | 112.35 |
These results agree fairly well with those obtained by the chloride and bromide methods. The second experiment is more trustworthy than the first. In it, we started with pure metal and the manipulations were so simple that no serious error could have been made in them. Hence it will only be necessary to consider the end-product, i.e., the cadmium sulphate. The titration showed that the sulphate was not basic owing to loss of sulphur trioxide, and after deducting the weight of the excess of sulphuric acid 92we must have left a weight of cadmium sulphate which is equivalent to the metal employed. The question now is, did it contain anything else and what would be its effect? Clearly the effect of water or any other impurity would be to lower the atomic weight found, hence the atomic weight must be at least as high as the experiment indicates. As the cadmium sulphate is deposited, at least the later part of it is from a strong sulphuric acid solution, it probably does not contain any water and in this case would fix a maximum value as well as the 93minimum value, and thus determine the atomic weight. It might be objected to the second experiment that the sulphuric acid found may have been present as SO3 and not as H2SO4 as was assumed. This seems highly improbable, and even if it were so the error introduced would be only about .03 of a unit in the atomic weight. As the first determination was found practically neutral, it does not apply to it at all. The most probable conclusion from these experiments is that the atomic weight of cadmium is about 112.35. A more thorough study of this method would have been made if time had permitted it.
As the chloride and bromide methods and the synthesis of cadmium sulphate all lead to approximately the same high result, it seemed probable that the oxide method which had given a much lower result (Morse & Jones 112.07) must be affected by some error. Accordingly it was examined in the manner about to be described. A set of crucibles was prepared as described by Morse and Jones in their work on this method, and in the present paper under the oxalate method. After they had been heated in a nickel crucible over 95a blast lamp and weighed, a weighed piece of cadmium was introduced into the smaller inside crucible, and dissolved in nitric acid with the aid of heat. An equal quantity of nitric acid was added to the tare. The acid was then evaporated off, and the resulting nitrate converted into oxide exactly as has already been described under the oxalate. The first experiment was made in this way and the second one exactly like it, only the porcelain crucible used was the one which had been employed in the first determination. The glaze had been removed by the 96cadmium oxide of the first determination, and before using for the second one the crucible was boiled out with nitric acid, and heated to constant weight over a blast lamp as before. Determinations III, IV and V were made in the same way except that the small inner crucible was platinum instead of porcelain. All weighings were reduced to the vacuum standard on the basis of 8.54 for the Sp. Gr. of cadmium and 8.15 for the Sp. Gr of cadmium oxide and 8.4 for the brass and 21 for the platinum weights.
The results are as follows:
| 97 | |||
| Cd | CdO | At. Wt. Cd. | |
|---|---|---|---|
| I. | 1.26142 | 1.44144 | 112.11 |
| II. | .99785 | 1.14035 | 112.04 |
| Average | 112.08 | ||
| III. | 1.11321 | 1.27247 | 111.84 |
| IV. | 1.02412 | 1.17054 | 111.91 |
| V. | 2.80966 | 3.21152 | 111.87 |
| Average | 111.87 | ||
The oxides resulting from these determinations were always tested for oxides of nitrogen, sometimes by using meta phenylene diamine and at other times by sulphanilic acid and naphthylamine sulphate, but no traces were ever found. The average of the determinations made in 98porcelain crucibles is 112.08. Morse and Jones obtained the same figure or, if their results are reduced to the vacuum standard, 112.06, by the same method under the same conditions. The results of the determinations made in platinum crucibles are equally constant, but their average is 111.88 being .20 of a unit lower. Therefore, more oxide is obtained when platinum crucibles are used instead of porcelain ones. In two cases the platinum crucibles were weighed at the end of the determinations after the cadmium oxide had been removed. Their weight remained unchanged. The most probable explanation 99of these facts seems to be that something is retained in the oxide in both cases, but that the amount is greater in the determination made in platinum crucibles than in those in which porcelain ones were employed. We should expect this, because in porcelain crucibles some of the oxide is absorbed forming a silicate, and any volatile impurity must be expelled from this part of the oxide. Not finding oxides of nitrogen, it was thought that gases probably nitrogen and oxygen might be occluded although Richards and Rogers (Amer. Chem. Jour. 15, 567.) had examined cadmium oxide prepared from the nitrate and found 100only a trace of gas. Accordingly two specimens of cadmium oxide obtained in the above determinations were powdered in an agate mortar and boiled with water for some time in order to remove any adhering air. They were then dissolved in dilute hydrochloric acid from which the air had been removed by boiling. A small amount of gas was found in each case but not nearly enough to account for the difference of .31 unit in the atomic weight of cadmium between 112.38 and the oxide method. In fact not more than about one sixth of the amount required was found. It may be that the powdering 101of the oxide and then boiling up in water may have been to severe a treatment, and that the greater part of the occluded gas escaped during these processes. It seems that there is at least some error due to occluded gases in methods involving the decomposition of cadmium nitrate to oxide, but no satisfactory idea of its magnitude could be obtained from these two experiments as carried out.
The following experiments were then made and they seem to give definite evidence not only of the existence of an error but also of its magnitude. Carbonate of cadmium was 102made by dissolving pure cadmium in nitric acid, adding an excess of ammonia and a small quantity of ammonium carbonate. After standing for some time the cadmium carbonate was filtered off and rejected. The filtrate was treated with an excess of ammonium carbonate and the precipitated cadmium carbonate allowed to digest in it for some time. After washing by decantation several times the carbonate was transferred to a funnel containing a porcelain filter-plate, covered with a piece of ashless filter paper of slightly larger diameter, and washed thoroughly. 103with water. It was then transferred to a platinum dish, care being taken to avoid contamination with filter paper and heated gently to convert it into oxide. The resulting oxide was powdered in an agate mortar, returned to the platinum dish and heated to incipient whiteness for seven hours in a muffle furnace. The temperature must not be too high, otherwise the oxide will distill readily leaving no residue. The oxide is slightly volatile at good red heat as was observed in trying to make a determinant at this temperature by the oxide method. 104A weighed portion of the oxide which had been prepared from the carbonate in the manner described was dissolved in a weighed porcelain crucible and the resulting nitrate converted into the oxide again by heat just as in the oxide method. This constitutes experiment I. Experiments two and three were made in exactly the same way except that a platinum crucible was used instead of a porcelain one. The results are:
| Initial Wt. | Final Wt. | Gain | Corresponding Error in At. Wt. | |
|---|---|---|---|---|
| I. | 2.95469 | 2.95650 | .00081 | −.24 |
| II. | 2.67717 | 2.67835 | .00117 | −.39 |
| III. | 3.00295 | 3.00422 | .00127 | −.38 |
105As we started with cadmium oxide, and, after passing to the nitrate, converted it back into the oxide, the weight should remain unchanged if the method is correct. However, this is not the case, but a large increase in weight takes place. The increase is larger in a platinum crucible than in a porcelain one, which accounts for the fact that a lower value for the atomic weight is found by the oxide method when they are used. The use of a porcelain crucible therefore diminishes the error, but does not eliminate it. The explanation of this has 106 already been given. The oxides obtained in these three experiments were tested for occluded gases in the manner already described, but only small amounts were found. Both of those made in platinum crucibles were tested for nitrate of cadmium with brucine and sulphuric acid with negative results. To show that the impurity was not converted into an ammonium salt when the oxide was dissolved in hydrochloric acid, a slight excess of caustic potash was added to the solution, the precipitate allowed to subside and the clean, supernatant liquid tested for 107ammonia with Nessler’s reagent. No ammonia was found. In order to make these experiments as severe a test as possible, a somewhat higher temperature was employed than had in the five experiments described under the oxide method. This was accomplished by boring out the stopcocks of the blast lamp so that a larger supply of gas was furnished. The two oxides in the platinum crucibles seemed to be constant in weight, but that in the porcelain crucible seemed to lose in weight slowly. The weight given was taken after four hours blasting, which is 108longer and at a higher temperature than was used in any of the five determinations made by the oxide method. If the cadmium oxide prepared from the carbonate retained any carbon dioxide, it would lose weight in being dissolved and reconverted into oxide. The above experiments therefore seem to furnish very strong evidence that there is an error of at least −.24 unit in the oxide method when porcelain crucibles are used and −.39 of a unit when platinum ones are employed. if .24 of a unit is added to 112.07 the result obtained when porcelain crucibles are used we get 112.31 109and adding .39 to 111.87 gives 112.26. Considering the small number of experiments made, the fact that they were made in such a way as to give a low value (numerically) for the error rather than a high one, and also that the error is probably variable to some extent, especially when porcelain crucibles are used, the corrected results agree as closely with 112.38, the average of the chloride, bromide and sulphate (synthetical) methods as could be expected. It must also be borne in mind that 112.38 is only to be regarded as an approximation to the atomic weight of cadmium. The increase in 110weight observed in converting the nitrate back into oxide might also be explained by assuming that the cadmium oxide used in the beginning of the experiments was richer in metal than the formula CdO indicated and that the increase in weight is due to this excess of metal being changed to oxide. The method of preparation of the oxide from the carbonate and the known properties of cadmium oxide render this view highly improbable, and the following two observations render it untenable:
1st. If this were the cause of the increase, the amount of increase would necessarily be 111the same in both platinum and porcelain crucibles, which is not the case.
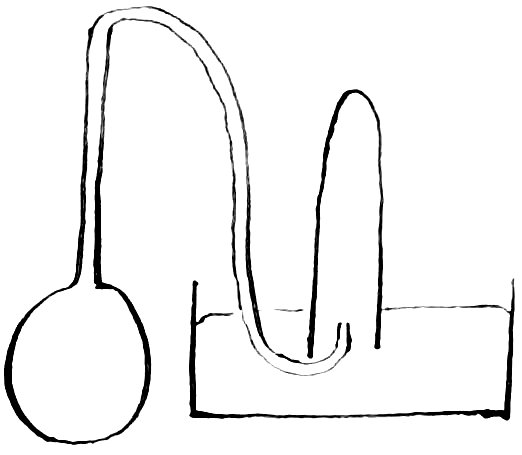
2nd. Three grammes of cadmium oxide made from the carbonate were dissolved in dilute hydrochloric acid from which the air had been expelled by boiling. The oxide, which is very compact, was placed in a glass bulb which had been blown at the end of a tube. After displacing the air by filling the entire apparatus with recently boiled water, the exit of the tube was placed under boiling dilute hydrochloric acid, and the bulb heated until the water boiled. It was then turned over so that the steam displaced nearly all the water. On removing the flame the dilute hydrochloric acid at once filled the bulb. The exit tube was then quickly placed under a narrow tube filled with mercury and inverted over mercury in a dish. The bulb was then heated until the oxide had dissolved. By this method the gas would be boiled out of the solution and collected in the top of the narrow tube. As only a very small amount of steam and dilute hydrochloric acid go over at the same time, there is no danger of the gas 113formed being absorbed to any considerable extent. It is well to put the oxide into the bulb before the tube is bend. If the hydrochloric acid is too strong, it must be cooled before entering the bulb as otherwise the reaction is too violent, and the experiment may be lost. This experiment shows that there is no excess of cadmium present in the oxide employed for no gas was found. If three grammes of the oxide contained enough metal to take up .00126 grms. of oxygen, .00016 grms of hydrogen should have been set free, and its volume under ordinary conditions of temperature and pressure would have been about 1.9 114cubic centimetres. This experiment would also have shown the presence of carbon dioxide if any had been present.
After having done the work which has just been described, we are in a position to turn to the oxalate method, which is the first method described in this paper. It involves the decomposition of cadmium nitrate, and is therefore affected by an error from this source, only it is not as large as in case of the oxide method. If 2.95650 grammes of 115cadmium oxide prepared in a porcelain crucible contain .00081 grammes of impurity, an error of −.24 of a unit would be introduced in the atomic weight as determined by the oxide method or +.10 in case the oxalate method were employed. That is the oxalate should give about 112.48 for the atomic weight of cadmium, but it really gives a very much lower result. Morse and Jones obtained 112.04 ± .035 by it, while Partridge obtained 111.81 ± .035 by it. If we take 112.38 for the atomic weight of cadmium, there appears to be a second error of .44 of a unit in the method as used by Morse and Jones, while 116Partridge’s result indicates an error of .57 of a unit. Partridge only moistened the oxide obtained from the oxalate with a few drops of nitric acid before making the final heating, and it seems probable therefore that he made no appreciable error on account of the final oxide retaining products of decomposition from cadmium nitrate. The most probable cause of this large error seems probably to be incomplete dehydration of the oxalate, or reduction to metal during the decomposition of the oxalate, and subsequent volatilization of some of it, or a combination of both of 117these. The nine determinations given in the earlier part of this paper of course vary so much that they are of no value whatever in determining the atomic weight. The reason that the first four are low is probably due in part to sublimation of cadmium, for on dissolving the resulting oxide in nitric acid a considerable quantity of metal was noticed in each case. In the others, the temperature was kept lower, and the decomposition took a longer time. No metal was observed on taking up in nitric acid. To be certain of what the cause of error is would 118require some very carefully conducted experiments, but as there are a number of much more reliable methods for determining the atomic weight of cadmium, it does not seem desirable to spend the time required in making them. It should be mentioned that Lenssen, in 1860, first employed this method. He made three determinations. 1.5697 grms of cadmium oxalate giving 1.0047 grammes of oxide, which gives a value of 112.043 for the atomic weight of cadmium . The difference between the highest and lowest determination was .391 of a unit.
A great deal of time was spent in trying to effect a partial synthesis of cadmium bromide in exactly the same manner as had been used in case of cadmium sulphate. No results were obtained because cadmium bromide is slowly volatile at 150°C, the temperature used, and retained some hydrobromic acid ever after more than 100 hours of drying. Some work was done in trying to establish the ratio between silver and Cadmium by dropping a weighed piece of cadmium into a solution of 120silver sulphate, the reaction being:
Silver nitrate cannot be used because it becomes reduced to nitrate even at a temperature of 0°C., as was shown by its reducing action on potassium permanganate, and by the reaction with meta-diamido benzene after the reaction had been completed. The main difficulty with the method is that air must be excluded in order to prevent oxidation and solution of some of the precipitated silver. The silver is perfectly free from cadmium if an excess of silver sulphate is used and the precipitated metal digested 121with it for some time. Since this part of the work was done, a paper by Mylius and Fromm (Ber. 1894, 630) appeared in which one of the reactions studied was that of cadmium on silver sulphate. They also found the resulting silver free from cadmium. The method seems very promising, but the work had to be discontinued for lack of time.
I. The work on the oxalate and sulphide methods described in this paper is of no value for determining the atomic weight of cadmium. It does not even enable us to fix an approximate value.
II. There are a number of errors in the chloride and bromide methods as they were used in this work, but they are not very large and partially compensate each other. Their results, 112.383 and 112.396 respectively, may be regarded as approximations to the true value.
III. The synthesis of cadmium sulphate as carried out is of especial value 123in fixing a minimum value for the atomic weight of cadmium. The result is 112.35, agreeing closely with that obtained by the bromide and chloride methods.
IV. There is an error in the oxide method due to products of decomposition of the nitrate being retained. Direct experiments gave .39 of a unit for this when platinum crucibles were used and .24 of a unit when porcelain ones were used. The calculated errors for porcelain and platinum crucibles are .30 and .51 of a unit respectively, if 112.38 is assumed as the atomic weight of Cadmium.
V. The average of the chloride, bromide, and sulphate methods 124is 112.38. This result is to be regarded as tentative and not as final since the main object of this work has been to find the cause of the discrepancy in some methods employed in determining this constant, rather than to make an atomic weight determination.
John Emery Bucher was born near Hanover, Pa., August 17, 1872. He entered Lehigh University in 1888 and graduated in 1891. During the past three years he has been a graduate student in the Johns Hopkins University.
Subjects: Chemistry, Mineralogy and Mathematics.